ccImpute algorithm to impute dropout events in the single-cell RNA-seq data
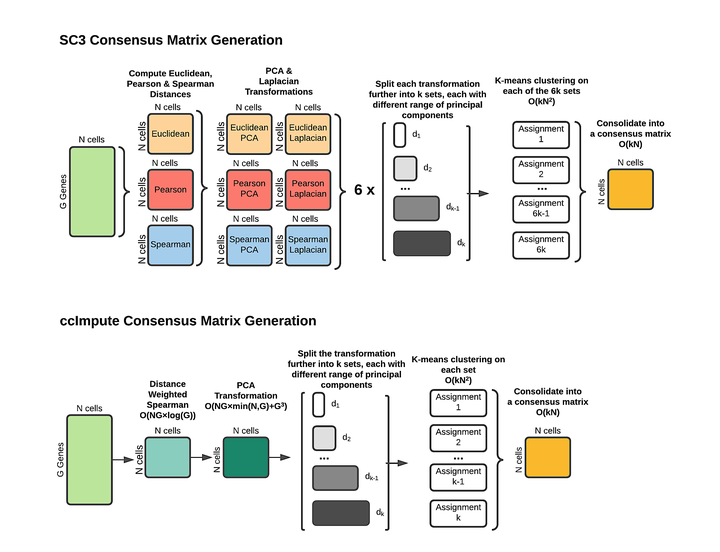
In recent years, the introduction of single-cell RNA sequencing (scRNA-seq) has enabled the analysis of a cell’s transcriptome at an unprecedented granularity and processing speed. The experimental outcome of applying this technology is a M by N matrix containing aggregated mRNA expression counts of M genes and N cell samples. From this matrix, scientists can study how cell protein synthesis changes in response to various factors, for example, disease versus non-disease states in response to a treatment protocol. This technology’s critical challenge is detecting and accurately recording lowly expressed genes. As a result, low expression levels tend to be missed and recorded as zero - an event known as dropout. This makes the lowly expressed genes indistinguishable from true zero expression and different than the low expression present in cells of the same type. This issue makes any subsequent downstream analysis difficult. To address this problem, we propose an approach to measure cell similarity using consensus clustering and demonstrate an effective and efficient algorithm that takes advantage of this new similarity measure to impute the most probable dropout events in the scRNA-seq datasets. We demonstrate that our approach exceeds the performance of existing imputation approaches while introducing the least amount of new noise as measured by clustering performance characteristics on datasets with known cell identities.
Dr. Hasan Kurban
Computer & Data Scientist
As a Computer Scientist and Machine Learning Researcher, I am passionate about developing intelligent systems that leverage data-driven approaches to address real-world challenges.